Osteogénesis Imperfecta
Definición:
La osteogénesis imperfecta (enfermedad de los huesos frágiles), causa más frecuente de osteoporosis hereditaria, es un trastorno generalizado del tejido conjuntivo debido a defectos del colágeno de tipo I.
Es una enfermedad congénita que se caracteriza porque los huesos de las personas que la sufren se rompen muy fácilmente, con frecuencia tras un traumatismo mínimo e incluso sin causa aparente. El espectro de la Osteogénesis Imperfecta es sumamente amplio, y abarca desde una forma mortal en el periodo perinatal hasta una forma leve cuyo diagnóstico puede ser dudoso o ambiguo en el adulto.
Frecuencia:
Es difícil determinar el número de personas afectadas por la enfermedad, ya que no hay censos donde consten semejantes datos. Además, podemos partir de la base de que hay gran cantidad de afectados con síntomas muy leves que no han sido ni serán nunca diagnosticados.
Se afirma que la incidencia es de uno entre veinte o treinta mil nacimientos (OIF), y otras según las cuales la cifra debe de andar por un afectado entre cada diez mil nacidos, incluyendo los casos que son diagnosticados a posteriori.
Historia:
Osteogénesis Imperfecta fue un término difundido por Vrolik (1849) para designar este síndrome congénito de naturaleza genética, y de presentación variable, caracterizado por fragilidad ósea, osteoporosis y fracturas. Ekman en 1788 lo denominó osteomalacia congénita, Lobstein en 1853 osteopsatirosis, Eddowes en 1900 síndrome de las escleróticas azules, Porak-Durante distrofia periostal o síndrome de Van Der Hoeve (sordera en 1917).
Originalmente la enfermedad estaba dividida en dos tipos: Osteogénesis Imperfecta Congénita y Osteogénesis Imperfecta Tardía. Esta clasificación fue superada por investigaciones realizadas por el Doctor Sillence quién la dividió en cuatro tipos:
Tipo I: es el tipo más frecuente, de transmite como autosomal dominante pero también puede ser el resultado de una mutación espontánea.
Tipo II: abarca aproximadamente el 10% de las personas afectadas. Resulta de una nueva mutación y es la forma más severa que de Osteogénesis Imperfecta.
Tipo III: abarca el 20%. Los enfermos sufren con frecuencia fracturas espontáneas.
Tipo IV: es de leve a moderado. La mayoría de las fracturas se presentan durante la infancia.
Patogenia:
Abarca todos los grupos raciales y étnicos. Este trastorno óseo generalmente se presenta en el nacimiento como una enfermedad hereditaria. La Osteogénesis Imperfecta se clasifica en cuatro grandes tipos (y otros subtipos), todas ellas son ocasionadas por defectos en la cantidad o estructura del colágeno Tipo 1, el cual es una parte importante de la matriz del hueso. El problema con el colágeno usualmente resulta de un defecto genético dominante que puede ser adquirido por diversos y diferentes mecanismos:
El defecto puede ser heredado en un patrón autosómico dominante de un padre afectado. Esto significa que un padre afectado que porta un gen único para este trastorno tiene un 50 % de posibilidades de tener hijos que lo padezcan y cualquier niño que lo herede resultará afectado.
El defecto puede adquirirse por una mutación espontánea que se presenta en el óvulo o espermatozoide individual que formó al niño. En este caso, ninguno de los padres porta el gen para el trastorno o está afectado por el mismo. Los padres, en este caso, no tienen más riesgo que la población general para tener otro hijo con dicho problema.
El defecto se puede adquirir a través de un patrón de herencia denominado mosaiquismo. Este fenómeno se presenta cuando un padre no está afectado, pero es portador de un porcentaje de espermatozoides u óvulos que portan el trastorno genético. Por lo tanto, aunque los padres no estén afectados, algunos de sus hijos pueden tener el trastorno y otros no. Se estima que más o menos del 2 al 7% de las familias no afectadas que han tenido un hijo con osteogénesis imperfecta tendrán otro hijo con esta enfermedad debido al fenómeno de mosaiquismo.
Una de cada 20.000 personas padece Osteogénesis Imperfecta. Una de cada 50.000 a 60.000 personas desarrolla las formas más graves de la enfermedad.
Todos los tipos de Osteogénesis Imperfecta se deben a defectos cualitativos o cuantitativos del colágeno de tipo I (principal componente de la matriz extracelular del hueso y la piel).
Debido al alto predominio del colágeno en el hueso se produce una desmineralización ósea anormal, pero también a otros niveles: escleróticas, piel, dientes, oídos, etc.
Causas:
La osteogénesis imperfecta se debe a la insuficiente y/o defectuosa formación del colágeno del cuerpo, como consecuencia de un fallo genético. El colágeno es una proteína de los tejidos y su función en la formación de los huesos se puede comparar a la de las nervaduras metálicas en torno a las cuales se monta la estructura de hormigón de una viga. Si la nervadura no es fuerte o no existe, la pieza de hormigón no adquirirá la forma adecuada o será sumamente frágil.
Trasmisión:
En cuanto a la forma en que se transmite, los especialistas no acaban de ponerse de acuerdo. Hasta no hace mucho parecía claro que la forma de transmisión de los tipos I y IV era dominante (estos dos tipos se pueden estudiar en familias donde la enfermedad aparece a lo largo de generaciones). La opinión generalmente aceptada era hasta hace muy poco que tanto el tipo II como el tipo III son el resultado de una transmisión recesiva, pero los más recientes estudios parecen reflejar dudas fundadas sobre esta generalización y apuntan a la posibilidad de que el mosaicismo (error genético en las células germinales de los progenitores) desempeñe un papel importante en la transmisión de los casos más graves de OI.
Además, la enfermedad también puede ser el resultado de una mutación espontánea y aparecer en familias sin ningún antecedente. En algunos casos, lo que puede parecer una mutación espontánea no lo es: hay afectados con síntomas tan leves que ignoran su condición hasta que tienen hijos con la enfermedad.
En cualquier caso, está claro que los afectados por osteogénesis imperfecta tienen un cincuenta por ciento de probabilidades de transmitirla a su descendencia.
Clasificación:
Se conocen varios tipos de la enfermedad, y su variación es muy grande de un individuo a otro. Incluso dentro del mismo tipo, puede haber personas con una mayor o menor impregnación.
Por decirlo con un ejemplo práctico, algunos pacientes sufren diez fracturas a lo largo de su vida, en tanto que otros pueden llegar a tener varios cientos de ellas.
El Dr. Sillence, una de las mayores autoridades mundiales en materia de Osteogénesis Imperfecta, estableció hace ya algunos años una clasificación de la misma en cuatro tipos: Tipo I, Tipo II, Tipo III y Tipo IV. Las clasificaciones no siempre son exactas, hay gente que comparte características de los tipos III/IV, otros que se definen como II/III, y otros que simplemente quedan fuera.
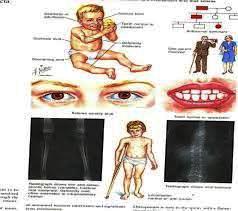
Síntomas:
Están compuestos por una triada:
• Fragilidad ósea.
• Escleróticas azules.
• Sordera prematura.
En los diversos tipos de Osteogénesis Imperfecta se puede encontrar una variedad de síntomas:
Fractura ósea:
• Presencia de más de un hueso fracturado en un sólo episodio (múltiple).
• Presente en el nacimiento.
• Después de un trauma menor.
• Deformidad de las extremidades o extremidades cortas.
• Sordera (la pérdida de la audición conductiva se puede presentar en adolescentes y adultos).
• Cifosis.
• Cifoescoliosis.
• Baja estatura.
• Deformidades dentales.
• Puente nasal bajo.
• Tórax en quilla.
• Tórax excavado.
• Pie plano.
• Laxitud de las articulaciones.
• Hipermovilidad.
• Piernas en arco.
• Huesos Vormianos (pequeños osículos dentro de las líneas de sutura craneana, que se pueden percibir en las radiografías del cráneo).
Otras manifestaciones:
• Tendencia a la formación de hematomas.
• Voz aguda.
• Estreñimiento.
• Sudoración excesiva.
• Músculos débiles.
• Rostro en forma triangular.
Sordera:
La pérdida significativa del sentido del oído se da aproximadamente en el 50% de las personas con Osteogénesis Imperfecta. En el caso de Osteogénesis Imperfecta tipo I, la forma más frecuente de aparición de pérdida del oído, comienza alrededor del los 20 – 30 años.
Hay tres tipos principales de alteración:
1-Conducción: resultado de un problema físico en el oído medio o externo; puede ser consecuencia de una infección del oído, obstrucción o por fractura de la cadena de huesecillos que se encuentran en el oído, por dentro del yunque.
2-Neurosensorial: cuando el oído interno no transmite la señal de sonido de forma adecuada al cerebro.
3-Mezcla: cuando están implicados el oído medio e interno.
La pérdida del oído también se puede clasificar según el grado de severidad (leve, moderada, severa y profunda) o por la frecuencia afectada (bajo, alto o todas las frecuencias).
Los síntomas que podemos encontrar son:
• Dificultad para entender ciertas palabras o partes de palabras.
• Preguntas frecuentes al interlocutor para que éste repita las palabras.
• Dificultad para entender por teléfono.
• Volumen excesivo de la televisión o la radio.
• Sensación de entorno ruidoso.
• Si el mal progresa, la pérdida del sentido del oído puede interferir con la comunicación normal, el rendimiento en el trabajo y en las actividades sociales y personales. Si no se trata adecuadamente puede provocar aislamiento y depresión.
Osteogénesis Imperfecta TIPO I (leve):
Presenta la tríada clínica en el 30-60% de los casos. A su vez la tipo I se subdivide en A o B según falte o exista dentinogénesis imperfecta, dientes descoloridos y frágiles debidos a la defectuosa formación de la dentina, que conlleva la decoloración y la fragilidad de los dientes. Además puede presentar facilidad para sangrar con los roces, laxitud articular, estatura baja comparado con otros familiares. Las fracturas son secundarias a traumatismos y disminuyen con la pubertad. Los huesos más afectados son los huesos largos que se rompen con facilidad aunque, consolidan en el tiempo adecuado con grandes callos fracturarios (a veces confundidos con tumores debido a su tamaño) y que no protegen contra nuevas fracturas en ese punto por lo que, hay que extremar las precauciones ya que, un movimiento mal controlado puede provocar una nueva fractura. Además las múltiples fracturas dan lugar con el tiempo a un arqueamiento de los huesos de las extremidades.
Dientes descoloridos y frágiles. La defectuosa formación de la dentina, que conlleva la decoloración y la fragilidad de los dientes
Osteogénesis Imperfecta TIPO II (letal):
Este tipo se hereda con carácter autosómico recesivo aunque también ocurre de forma esporádica. Es la forma más grave y también recibe el nombre de Osteogénesis Imperfecta congénita. Aparece en el momento del nacimiento, los niños afectados presentan numerosas fracturas incapacitantes producidas en el mismo útero o debidas a las maniobras del parto, mostrando al examen radiológico “huesos arrugados”.
Hay intensa micromielia e incurvación de los miembros, las piernas están en abducción y forman un ángulo recto con el cuerpo, que adopta una postura en “piernas de rana”. Normalmente conduce a la muerte en poco tiempo (nacen muertos o fallecen a las pocas horas). Los neonatos afectados son de baja estatura y presentan severas deformidades de las extremidades, éstas generalmente cortas aunque la longitud de las manos y pies es normal. Presentan escleróticas gris azulado oscuro, la piel muestra una estriación transversal o pliegues, el cráneo generalmente grande se palpa blando por la defectuosa o nula osificación, de consistencia elástica llamado por esto “cráneo en pelota de goma”, no obstante, el desarrollo mental es normal. Las suturas y fontanelas son amplias pero la proporción entre la base del cráneo y la calota está conservada.
Osteogénesis Imperfecta TIPO III (deformante progresiva):
Representa la forma progresiva y deformante de la enfermedad (no mortal), se caracteriza por numerosas fracturas óseas retraso en el crecimiento y severas deformaciones del esqueleto. Esta forma se hereda con carácter autosómico recesivo. El peso y longitud al nacer suelen ser inferiores a lo normal. Las fracturas se encuentran presentes al nacer (los huesos son menos frágiles que en el Tipo II). Debido a la desorganización de la matriz ósea, las radiografías de las metáfisis producen una imagen en “palomitas de maíz”. Si bien las escleróticas son azules al nacer se tornan blancas. Las alteraciones dentarias son comunes.
Osteogénesis Imperfecta TIPO IV (moderadamente severa):
Se hereda con carácter autosómico dominante similar al Tipo I pero, con escleróticas normales. Estos pacientes nacen con fracturas e incurvaciones de los huesos largos de los miembros inferiores. Esta enfermedad parece ser heterogénea y puede asociarse a alteraciones en los dientes (tipo IV A) o no (tipo IV B). El colágeno anormal no permite la maduración de la cortical ósea de modo que, la cortical está compuesta por hueso primitivo y pequeñas áreas de hueso laminar, con los años (adolescencia o más tarde) la cortical madura. Las radiografías revelan osteoporosis, ensanchamiento metafisario y compresiones vertebrales. Son tratados con medidas ortopédicas y rehabilitación, presentan una disminución del colágeno Tipo I, muestran dentinogénesis imperfecta, escleróticas blancas y no hay sordera. Suele presentar cifoescoliosis y laxitud ligamentosa.
Diagnóstico:
El diagnóstico se hace por medio de estudios de colágeno que se realizan con una biopsia de perforación. Se aprecia una disminución del colágeno Tipo I (que forma las laminillas óseas a nivel de la piel) y mayor proporción del colágeno Tipo III, en todos los tipos de osteogénesis imperfecta. Los análisis bioquímicos suelen dar resultados normales aunque, muchas veces se descubre una hiperfosfatasemia sobretodo del tipo ácido y un aumento en la excreción urinaria de Mucopolisacaridosis e hidroxiprolona pero carece de significación para el diagnóstico diferencial, pues son síntomas secundarios de osteopatía.
Una vez conocido el diagnóstico molecular específico, a los miembros de la familia se les puede hacer una prueba por medio de un examen de sangre para ADN.
En la mayoría de las personas afectadas de OI, los rayos x se convierten en una prueba frecuente y necesaria de asistencia al diagnóstico y tratamiento. Hay peligro para la salud con la exposición frecuente a los rayos x, directamente relacionada con la intensidad de éstos. Es conveniente guardar un registro con las fechas en las que se le han realizado radiografías.
Diagnóstico prenatal:
La prueba de ADN en muestras prenatales de vellosidades coriónicas puede ayudar a hacer el diagnóstico durante el embarazo. La Osteogénesis Imperfecta severa se puede detectar por medio de un ultrasonido prenatal a las 16 semanas de gestación.
Tratamiento:
La OI no se puede paliar, por tanto, suministrando calcio a los enfermos. No existe hasta el presente ninguna forma de inducir a las células del cuerpo a producir más colágeno o producir colágeno de calidad.
Cuando hablamos de terapia de la osteogénesis imperfecta nos estamos refiriendo a tratamientos correctivos y paliativos de la enfermedad, no a una curación. Hasta ahora no se ha hallado ningún método para inducir a las células a mejorar o incrementar su producción de colágeno.
En este sentido, hay tres aspectos terapéuticos interesantes: la fisioterapia, el correcto tratamiento de las fracturas y la introducción de agujas telescópicas Bailey en los huesos largos para prevenir curvaturas y fracturas.
Mención aparte merece el empleo de diferentes sustancias que ayudan a incrementar la densidad ósea, bifosfonatos y pamidronato; así como también el trasplante de médula ósea. Durante estos últimos años se están llevando a cabo en todo el mundo experimentos en este campo y los resultados parecen ser muy prometedores.
Las fracturas se deben reparar rápidamente de manera convencional para evitar deformidades. No existe un tratamiento específico para la enfermedad subyacente. Sin embargo, existen diversas terapias que pueden mejorar la calidad de vida en pacientes que tienen Osteogénesis Imperfecta:
La buena nutrición y el ejercicio supervisado son puntos claves para ayudar a optimizar la fortaleza ósea y muscular. La fisioterapia y la rehabilitación pueden ser muy beneficiosas. La natación es un excelente ejercicio para ponerse en forma en muchos pacientes con Osteogénesis Imperfecta.
Los procedimientos quirúrgicos como el implante de varillas metálicas en los huesos pueden ayudar a su fortalecimiento y a prevenir deformidades. El uso de bragueros y de ayudas para la marcha es útil en algunas personas.
El uso de bifosfonatos en los niños con Osteogénesis Imperfecta se está investigando en la actualidad con algunos resultados prometedores. Otras intervenciones médicas que incluyen el trasplante de médula ósea, el uso de la hormona del crecimiento y la terapia genética también se están investigando.
Los bifosfonatos tienen una influencia directa sobre el metabolismo del hueso: inhiben la actividad de las células que “eliminan” hueso. De esa forma, el organismo produce más masa ósea de la que pierde, y lógicamente, el hueso se hace más grueso, por lo tanto se hace más estable y menos propenso a las fracturas. Otro de los efectos colaterales de este tratamiento es la desaparición de los dolores crónicos que suelen afectar a estos pacientes. Un efecto que se ha observado en niños es el aumento del apetito, el incremento de la actividad y, en general, una considerable mejora en el estado general del paciente.
Hay diferentes tipos de bifosfonatos: el pamidronato, el alendronato y recientemente, el zolendronato, que aún se encuentra en fase de prueba y del que se esperan resultados muy prometedores.
El pamidronato tiene la propiedad de “adherirse” al hueso y de no acumularse en ningún tejido blando (corazón, riñón, pulmón, hígado, cerebro, etc.). Todo el pamidronato que no se depositó en el esqueleto es eliminado del organismo a las pocas horas de haber sido inyectado o tomado por boca. Por ello, es una medicación considerada muy segura por las autoridades sanitarias, ya que resulta difícil que provoque malestar secundario. En el hueso, el pamidronato se concentra en los sitios de destrucción o de recambio del mineral y paraliza al osteoclasto. No afecta adversamente al osteoblasto, por el contrario, estudios muy nuevos indican que hasta podría mejorarse la vitalidad del mismo.
Al cabo de cierto tiempo de tratamiento, se consigue alcanzar un balance positivo a favor del aumento de mineral en todo el esqueleto por eso el hueso se calcifica más. Los resultados pueden variar según el tipo de Osteogénesis Imperfecta, el tipo de metabolismo óseo, el período de crecimiento del paciente, el grado de movilidad del mismo, su alimentación, etc. Estos son factores que hacen que la experiencia con pamidronato en un niño no sea igual en otro.
Los trasplantes de médula ósea tal como se realizan en la actualidad, no constituyen una cura, pero si se realizan con suficiente anticipación, existe la esperanza de que disminuyan o hasta evitarán los síntomas de la OI. Se está dirigiendo un ensayo clínico de trasplante de médula ósea mediante el cual se realiza un trabajo de ingeniería en la médula, y se investiga si se puede mejorar el desenlace en los niños. Durante los próximos años, analizarán de qué manera este nuevo trasplante de médula ósea es positivo para los niños, y cuánto tiempo pueden durar los beneficios de esos trasplantes.
Complicaciones:
• Neumonía recurrente.
• Falla cardíaca (cor pulmonale).
• Lesión cerebral.
• Deformidad permanente.
• Problemas de respiración.
• Pérdida de la audición.
Consideraciones obstétricas:
• Las complicaciones que se pueden presentar son:
• Compromiso respiratorio en especial en mujeres de talla muy baja y con escoliosis asociada.
• Desproporción céfalo-pélvica.
• Diastasis de la sínfisis pubiana.
La forma de terminación del embarazo debe ser por cesárea para evitar traumatismos del feto durante su pasaje por el canal de parto, excepto en los casos de Osteogénesis Imperfecta Tipo II. El parto por fórceps está contraindicado por el riesgo de producir fracturas craneales y hemorragia intracraneal. El uso de anestesia general se ha visto asociado a mayor riesgo de hipertermia maligna y las anestesias raquídeas y peridural pueden producir fracturas vertebrales en casos de severa osteopenia.
Pronóstico:
La osteogénesis imperfecta tiene un pronóstico muy variable, dependiendo del grado en que cada individuo esté afectado. La enfermedad en sí no es letal. Sin embargo, las personas afectadas por las formas más graves pueden tener importantes problemas colaterales.
Mientras que los afectados por tipos más leves (I y IV según la clasificación de Sillence), en general no tienen más complicaciones que las que impongan sus fracturas y deformaciones óseas, así como las intervenciones quirúrgicas que sean necesarias para tratarlas, la OI del tipo II (siempre según Sillence) suele revestir mucha gravedad y puede llegar a ser letal, debido a las hemorragias que causan las fracturas múltiples en el recién nacido.
El tipo III de Sillence tiene un pronóstico variable. En los casos en que la deformación ósea es grave y el volumen torácico es escaso, se pueden presentar problemas de ventilación, que en algunos casos pueden dar lugar a neumonías.
En todos los tipos se pueden presentar también problemas cardiovasculares de diferente pronóstico.
A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por OI es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal, dentro de las limitaciones que imponga el grado de movilidad de cada uno.
Se puede presentar deformidad permanente de las extremidades y puede ocurrir daño cerebral debido a las fracturas de cráneo. Dicho trastorno puede ser fatal y se clasifica según el tipo:
Tipo I. Compatible con una expectativa normal.
Tipo II. La mayoría de las personas afectadas, aunque no todas, mueren en etapas tempranas de la niñez.
Tipo III. Deformidad progresiva con reducción de la expectativa de vida.
Tipo IV. Compatible con una expectativa de vida normal.
En todos los tipos se pueden presentar también problemas cardiovasculares de diferente pronóstico.
A pesar de la deformación ósea y la frecuencia de fracturas, la longevidad de una persona afectada por Osteogénesis Imperfecta es igual a la de cualquier otra. Sus cualidades intelectuales no están mermadas de ninguna forma por la enfermedad y pueden llevar una vida normal, dentro de las limitaciones que imponga el grado de movilidad de cada uno.
En la actualidad a pesar de los avances tecnológicos, la radiología convencional continua siendo el pilar para el diagnóstico imagenológico de Osteogénesis Imperfecta, utilizándose sólo métodos complementarios para eventuales complicaciones.
Dr. Avilio Méndez Flores